- ACÉTYLACÉTIQUES (ESTER ET SYNTHÈSES)
- ACÉTYLACÉTIQUES (ESTER ET SYNTHÈSES)L’acide acétylacétique est le nom usuel du butanone-3-oïque:
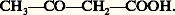 Cet acide a été isolé dans les urines pathologiques. Il peut être obtenu par une hydrolyse prudente de son ester éthylique. Ce dernier, beaucoup plus important, est un intermédiaire utilisé dans l’industrie pharmaceutique et dans celle des colorants. Il intervient dans la synthèse de nombreuses familles chimiques: pyridines, purines, furannes, pyrazoles, pyrroles, etc. Il présente une remarquable tautomérie céto-énolique (protomérie). L’acide est un solide très soluble dans l’eau et il est caractérisé par une très grande instabilité commune à tous les acides 廓-cétoniques énolisables: par chauffage, il se décarboxyle en énol de l’acétone (réaction 1). Sa capacité de réagir comme nucléophile carboné au niveau du carbone méthylénique central permet de l’utiliser pour introduire sur les substrats électrophiles le groupement acétyle CH3 漣C 略O.Préparation de l’ester éthyliqueLa méthode la plus classique de préparation est la condensation, selon Claisen, de l’ester énolisable acétate d’éthyle, en présence d’un catalyseur basique (éthylate ou amidure de sodium). La base forte arrache un proton à l’acétate d’éthyle pour former, dans une réaction équilibrée, le carbanion conjugué correspondant (la constante d’équilibre, dans le cas de l’éthylate, est d’environ 10-8). Celui-ci attaque une autre molécule d’acétate d’éthyle au niveau du carbone fonctionnel (centre électrophile) en expulsant une molécule d’éthylate de sodium. Ce dernier réagit comme une base vis-à-vis de l’ester acétylacétique formé en donnant, de façon réversible, un sel de sodium avec élimination d’éthanol (la constante d’équilibre est d’environ 103). Le premier équilibre est normalement défavorable à la formation du produit de condensation: on le déplace en éliminant l’éthanol par distillation en continu; la formation de l’énolate de sodium de l’ester 廓-cétonique favorise également ce déplacement. La base doit donc être employée en quantité stœchiométrique et l’ester acétylacétique est libéré de son sel par une hydrolyse acide douce (réactions 2). Industriellement, l’acétylacétate d’éthyle est préparé par addition de l’éthanol sur le dicétène (réaction 3).Tautomérie de l’acétylacétate d’éthyleL’ester acétylacétique est un liquide incolore, d’odeur agréable, assez soluble dans l’eau. Il se présente, à l’état pur et dans les conditions normales de température et pression, comme un mélange en équilibre de deux formes tautomères (réaction 4) renfermant 92 p. 100 de forme cétonique (I) et 8 p. 100 de forme énolique chélatée (II). Ce mélange est appelé allélotrope . La vitesse d’équilibration dépend de la température mais surtout de la présence de catalyseurs. Une trace d’acide ou de base accélère la transformation réversible des deux formes (réactions 5).En présence d’une trace d’acide, le catalyseur est l’acide conjugué (III) de la forme cétonique (réaction 5a); en présence d’une base, c’est l’anion énolate (IV) formé par arrachement du proton méthylénique acide (réaction 5b). En l’absence rigoureuse de catalyseur, la vitesse d’équilibration est relativement lente et l’on peut, par refroidissement brutal à 漣 78 0C d’une solution dans l’éther, obtenir la forme cétonique (I) cristallisée. La forme énolique peut être obtenue soit par distillation, sous pression très réduite dans une verrerie en silice (absence de l’alcalinité du verre), car elle est la plus volatile (chélation), soit par addition d’acide chlorhydrique gazeux et sec à une suspension, dans l’éther méthylique, du sel de sodium de l’ester: l’énolate (IV), anion ambident, se protone (acide dur) préférentiellement sur l’oxygène (base dure) plutôt que sur le carbone (base molle). Les solvants protiques favorisent la forme cétonique (99,5 p. 100 dans l’eau, 90 p. 100 dans l’éthanol) solvatable spécifiquement au niveau des fonctions carbonylées, tandis que, dans les solvants aprotiques, la forme énolique (interaction spécifique intramoléculaire) est un peu plus abondante (32 p. 100 dans le disulfure de carbone, 48 p. 100 dans l’hexane).Réactions du méthylène actifCe groupe méthylène porte un hydrogène acide (pK a = 11) qui peut être l’objet de réactions de substitution par les réactifs électrophiles, notamment par les halogènes, l’acide nitreux et les sels de diazonium; dans les trois cas, c’est la forme énolique qui intervient. Cette acidité se manifeste également par la formation, avec les cations des métaux de transition, de sels complexes dans lesquels deux anions énolate coordonnent le cation métallique. Le sel complexe formé par mélange de l’ester et d’une solution aqueuse d’acétate cuivrique a la structure plane donnée par la formule 6. Sa couleur n’est pas celle de l’ion cuivrique, il est peu soluble dans l’eau, et la potasse ne précipite pas l’hydroxyde cuivrique de cette solution qui ne conduit pas le courant électrique. Par contre, il est soluble dans les solvants organiques, cristallise bien et fond à point fixe.Synthèses acétylacétiques et autres réactionsL’anion énolate est un nucléophile ambident, mou au niveau du carbone, dur au niveau de l’oxygène. Ses réactions sont nombreuses: il est alkylé au carbone par les halogénures d’alkyle – électrophiles mous – (réaction 7a); l’ester alkylé obtenu peut réagir à nouveau dans les mêmes conditions et conduire au dérivé dialkylé (réaction 7b).L’hydrolyse acide de ces esters donne l’acide acétylacétique substitué qui se décarboxyle facilement par chauffage et conduit à une méthylcétone (VIII dans la réaction 7c).Par traitement basique (potasse alcoolique à l’ébullition), ces esters subissent une réaction de fragmentation (rétro Claisen) qui conduit à un acide 見-disubstitué (IX dans la réaction 7d).L’acylation de l’anion énolate par les chlorures d’acides (électrophiles plus durs que les halogénures d’alkyle) donne en général un mélange de dérivés acylés au carbone et acylés à l’oxygène.En présence d’un catalyseur basique, l’ester acétylacétique s’additionne sur les doubles liaisons activées par conjugaison avec un groupe électro-attracteur comme carbonyle, carboxyle, nitrile, nitro: c’est l’importante réaction de Michael. L’énolate formé par le catalyseur basique attaque le système conjugué. Le bilan de la réaction est une addition-1,2 sur la double liaison, alors que le mécanisme correspond à une addition-1,4 suivie d’une protomérie.L’anion énolate, formé par action d’une base assez forte comme la pipéridine sur l’ester acétylacétique, s’additionne sur les dérivés carbonylés en formant un alcool qui, s’il est secondaire ou tertiaire, se déshydrate en dérivé alkylidène (X dans la réaction 8).Réalisée en présence d’ammoniac, cette addition, qui conduit à un substrat éthylénique doublement conjugué (X), se poursuit par une addition de Michael de l’énamine nucléophile (XI) qui résulte de l’amination d’une deuxième molécule d’ester (réaction 9).La dihydropyridine (XII) ainsi obtenue, soumise à une hydrolyse acide et une oxydation (nitrique), donne finalement la triméthyl-2,4,6 pyridine (XIII, appelée couramment sym collidine). C’est la classique synthèse des pyridines de Hantzsch.D’autres hétérocycles sont également obtenus par réaction sur l’ester acétylacétique de nucléophiles azotés. C’est le cas de la N-phénylméthylpyrazolone (XIV), intermédiaire important en chimie pharmaceutique et en chimie photographique, qui résulte de la condensation de la phénylhydrazine (réaction 10).
Cet acide a été isolé dans les urines pathologiques. Il peut être obtenu par une hydrolyse prudente de son ester éthylique. Ce dernier, beaucoup plus important, est un intermédiaire utilisé dans l’industrie pharmaceutique et dans celle des colorants. Il intervient dans la synthèse de nombreuses familles chimiques: pyridines, purines, furannes, pyrazoles, pyrroles, etc. Il présente une remarquable tautomérie céto-énolique (protomérie). L’acide est un solide très soluble dans l’eau et il est caractérisé par une très grande instabilité commune à tous les acides 廓-cétoniques énolisables: par chauffage, il se décarboxyle en énol de l’acétone (réaction 1). Sa capacité de réagir comme nucléophile carboné au niveau du carbone méthylénique central permet de l’utiliser pour introduire sur les substrats électrophiles le groupement acétyle CH3 漣C 略O.Préparation de l’ester éthyliqueLa méthode la plus classique de préparation est la condensation, selon Claisen, de l’ester énolisable acétate d’éthyle, en présence d’un catalyseur basique (éthylate ou amidure de sodium). La base forte arrache un proton à l’acétate d’éthyle pour former, dans une réaction équilibrée, le carbanion conjugué correspondant (la constante d’équilibre, dans le cas de l’éthylate, est d’environ 10-8). Celui-ci attaque une autre molécule d’acétate d’éthyle au niveau du carbone fonctionnel (centre électrophile) en expulsant une molécule d’éthylate de sodium. Ce dernier réagit comme une base vis-à-vis de l’ester acétylacétique formé en donnant, de façon réversible, un sel de sodium avec élimination d’éthanol (la constante d’équilibre est d’environ 103). Le premier équilibre est normalement défavorable à la formation du produit de condensation: on le déplace en éliminant l’éthanol par distillation en continu; la formation de l’énolate de sodium de l’ester 廓-cétonique favorise également ce déplacement. La base doit donc être employée en quantité stœchiométrique et l’ester acétylacétique est libéré de son sel par une hydrolyse acide douce (réactions 2). Industriellement, l’acétylacétate d’éthyle est préparé par addition de l’éthanol sur le dicétène (réaction 3).Tautomérie de l’acétylacétate d’éthyleL’ester acétylacétique est un liquide incolore, d’odeur agréable, assez soluble dans l’eau. Il se présente, à l’état pur et dans les conditions normales de température et pression, comme un mélange en équilibre de deux formes tautomères (réaction 4) renfermant 92 p. 100 de forme cétonique (I) et 8 p. 100 de forme énolique chélatée (II). Ce mélange est appelé allélotrope . La vitesse d’équilibration dépend de la température mais surtout de la présence de catalyseurs. Une trace d’acide ou de base accélère la transformation réversible des deux formes (réactions 5).En présence d’une trace d’acide, le catalyseur est l’acide conjugué (III) de la forme cétonique (réaction 5a); en présence d’une base, c’est l’anion énolate (IV) formé par arrachement du proton méthylénique acide (réaction 5b). En l’absence rigoureuse de catalyseur, la vitesse d’équilibration est relativement lente et l’on peut, par refroidissement brutal à 漣 78 0C d’une solution dans l’éther, obtenir la forme cétonique (I) cristallisée. La forme énolique peut être obtenue soit par distillation, sous pression très réduite dans une verrerie en silice (absence de l’alcalinité du verre), car elle est la plus volatile (chélation), soit par addition d’acide chlorhydrique gazeux et sec à une suspension, dans l’éther méthylique, du sel de sodium de l’ester: l’énolate (IV), anion ambident, se protone (acide dur) préférentiellement sur l’oxygène (base dure) plutôt que sur le carbone (base molle). Les solvants protiques favorisent la forme cétonique (99,5 p. 100 dans l’eau, 90 p. 100 dans l’éthanol) solvatable spécifiquement au niveau des fonctions carbonylées, tandis que, dans les solvants aprotiques, la forme énolique (interaction spécifique intramoléculaire) est un peu plus abondante (32 p. 100 dans le disulfure de carbone, 48 p. 100 dans l’hexane).Réactions du méthylène actifCe groupe méthylène porte un hydrogène acide (pK a = 11) qui peut être l’objet de réactions de substitution par les réactifs électrophiles, notamment par les halogènes, l’acide nitreux et les sels de diazonium; dans les trois cas, c’est la forme énolique qui intervient. Cette acidité se manifeste également par la formation, avec les cations des métaux de transition, de sels complexes dans lesquels deux anions énolate coordonnent le cation métallique. Le sel complexe formé par mélange de l’ester et d’une solution aqueuse d’acétate cuivrique a la structure plane donnée par la formule 6. Sa couleur n’est pas celle de l’ion cuivrique, il est peu soluble dans l’eau, et la potasse ne précipite pas l’hydroxyde cuivrique de cette solution qui ne conduit pas le courant électrique. Par contre, il est soluble dans les solvants organiques, cristallise bien et fond à point fixe.Synthèses acétylacétiques et autres réactionsL’anion énolate est un nucléophile ambident, mou au niveau du carbone, dur au niveau de l’oxygène. Ses réactions sont nombreuses: il est alkylé au carbone par les halogénures d’alkyle – électrophiles mous – (réaction 7a); l’ester alkylé obtenu peut réagir à nouveau dans les mêmes conditions et conduire au dérivé dialkylé (réaction 7b).L’hydrolyse acide de ces esters donne l’acide acétylacétique substitué qui se décarboxyle facilement par chauffage et conduit à une méthylcétone (VIII dans la réaction 7c).Par traitement basique (potasse alcoolique à l’ébullition), ces esters subissent une réaction de fragmentation (rétro Claisen) qui conduit à un acide 見-disubstitué (IX dans la réaction 7d).L’acylation de l’anion énolate par les chlorures d’acides (électrophiles plus durs que les halogénures d’alkyle) donne en général un mélange de dérivés acylés au carbone et acylés à l’oxygène.En présence d’un catalyseur basique, l’ester acétylacétique s’additionne sur les doubles liaisons activées par conjugaison avec un groupe électro-attracteur comme carbonyle, carboxyle, nitrile, nitro: c’est l’importante réaction de Michael. L’énolate formé par le catalyseur basique attaque le système conjugué. Le bilan de la réaction est une addition-1,2 sur la double liaison, alors que le mécanisme correspond à une addition-1,4 suivie d’une protomérie.L’anion énolate, formé par action d’une base assez forte comme la pipéridine sur l’ester acétylacétique, s’additionne sur les dérivés carbonylés en formant un alcool qui, s’il est secondaire ou tertiaire, se déshydrate en dérivé alkylidène (X dans la réaction 8).Réalisée en présence d’ammoniac, cette addition, qui conduit à un substrat éthylénique doublement conjugué (X), se poursuit par une addition de Michael de l’énamine nucléophile (XI) qui résulte de l’amination d’une deuxième molécule d’ester (réaction 9).La dihydropyridine (XII) ainsi obtenue, soumise à une hydrolyse acide et une oxydation (nitrique), donne finalement la triméthyl-2,4,6 pyridine (XIII, appelée couramment sym collidine). C’est la classique synthèse des pyridines de Hantzsch.D’autres hétérocycles sont également obtenus par réaction sur l’ester acétylacétique de nucléophiles azotés. C’est le cas de la N-phénylméthylpyrazolone (XIV), intermédiaire important en chimie pharmaceutique et en chimie photographique, qui résulte de la condensation de la phénylhydrazine (réaction 10).
Encyclopédie Universelle. 2012.